Pluripotent stem cells can give rise to all cells and constitute the mature organism. These specialized cells can be cultured in vitro and are referred to as embryonic stem cells (ESCs). ESCs have undergone a great revolution in developmental biology. These in vitro grown ESCs exhibit the potential to generate all lineages of the embryo in vivo and can give rise to any type of somatic cells such as cardiomyocytes, smooth muscle cells, endothelial cells, neuronal cells and hepatocytes upon in vitro differentiation. The advantages of embryonic stem (ESCs) cells over other cell types are their accessibility to genetic manipulation. They can easily undergo genetic modifications while remaining pluripotent, and can be selectively propagated, allowing the clonal expansion of genetically altered cells in culture. Human ESCs gained popularity as a valuable cellular source for the treatment of many degenerative diseases such as ischemic heart failure, Parkinson’s disease, Alzheimer’s disease, diabetes, spinal cord injuries and age-related macular degeneration. In 2010, first time human ESCs were employed to treat spinal cord injuries and there after a dozen clinical trials have been conducted with human ESCs to treat severe ischemic left ventricular dysfunction, age-related macular degeneration, Parkinson’s disease and diabetes, among other degenerative conditions.
However, the human ESC-based clinical trials suffer immensely from the ethical concerns regarding the use of cells of embryonic origin as well as from failed in vitro fertilized embryos that could result in abnormal development, and from the concerns of immune rejection after transplantation due to the allogenic origin of ESCs. Takahashi and Yamanaka (2016) made a breakthrough discovery of reprograming somatic cells to pluripotent state. In their study, somatic cells such as skin biopsy derived fibroblasts and peripheral blood derived T lymphocytes were reprogrammed through the forced ectopic expression of the transcriptional factors OCT4, SOX2, KLF, c-MYC, NANOG and LIN28. These cells, termed as induced pluripotent stem cells (iPSCs), exhibit similar gene expression, epigenetic profile and the differentiation potential to give rise to any type of somatic cells as that of ESCs.
ESCs and iPSCs have wide application in Bio-medical research.
1. Basic Research: Understanding cell fate control, Cell rejuvenation, Studying pluripotency, Tissue and organ development and Physiology.
2. Drug Discovery: Drug discovery for cardiovascular disease, Drug discovery for neurological and neuropsychiatric diseases, Drug discovery for rare diseases etc.
3. Toxicology Studies: Relative use of iPSC-derived cell types in toxicity testing.
4. Disease Modeling: Cardiovascular diseases model, Percent share utilization of iPSC for cardiovascular disease modeling, Proportion of iPSC Sources in cardiac studies, Proportion of vector types used in reprogramming, Proportion of differentiated cardiomyocytes used in disease modeling, iPSC-derived organoids for modeling development and disease, Modeling liver diseases using iPSC-derived Hepatocytes, iPSCs in neurodegenerative disease modeling, and Cancer-derived iPSCs.
5. Cell-Based Therapies: Cell therapy for AMD, Autologous iPSC-RPE for AMD, Allogeneic iPSC-RPE for AMD, iPSC-derived dopaminergic neurons for Parkinson's disease, iPSC-derived NK cells for solid cancers, iPSC-derived cells for GvHD, iPSC-derived cells for spinal cord injury, iPSC-derived cardiomyocytes for ischemic cardiomyopathy.
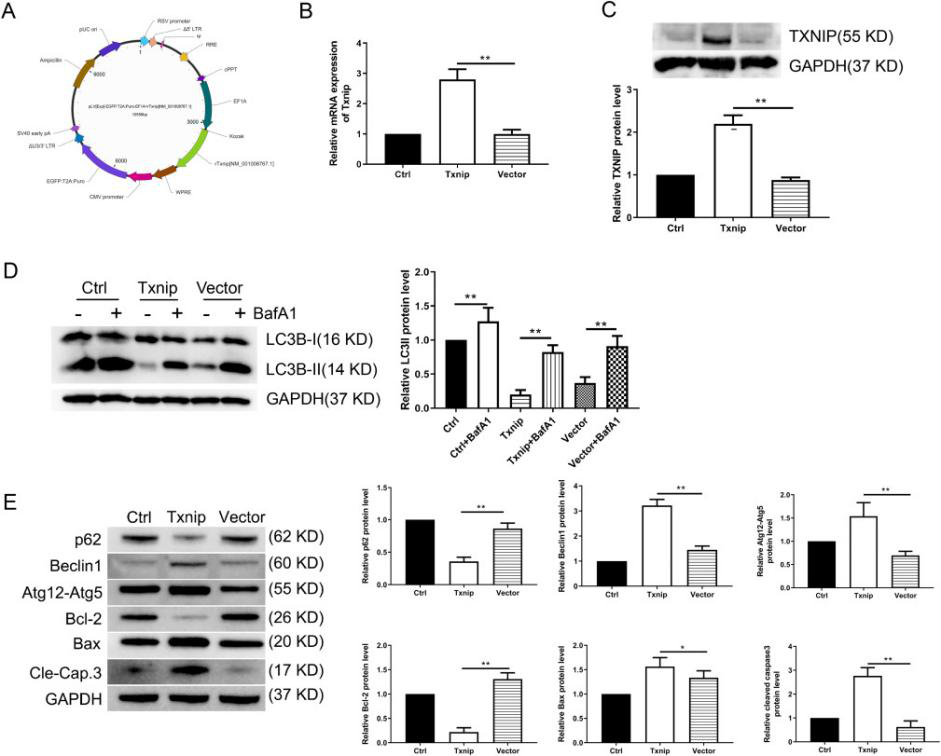
The CRISPR system, a potent system for genome editing, used for gene knockout or knock-in genome manipulations through substitution of a target genetic sequence with a desired donor sequence. The CRISPR system combined with inducible pluripotent stem cells can generate single or multiple gene knockouts, correct mutations, or insert reporter transgenes. ESCs and iPSCs were engineered with CRISPR used to explore genetic determinants of lineage choice, differentiation, and stem cell fate, allowing investigators to study how various genes or noncoding elements contribute to specific processes and pathways. Genome editing in ESCs and iPSCs cellular models can advance research program in the area of functional genomics, signaling pathways, drug discovery, drug response, cancer research and cell therapy etc.
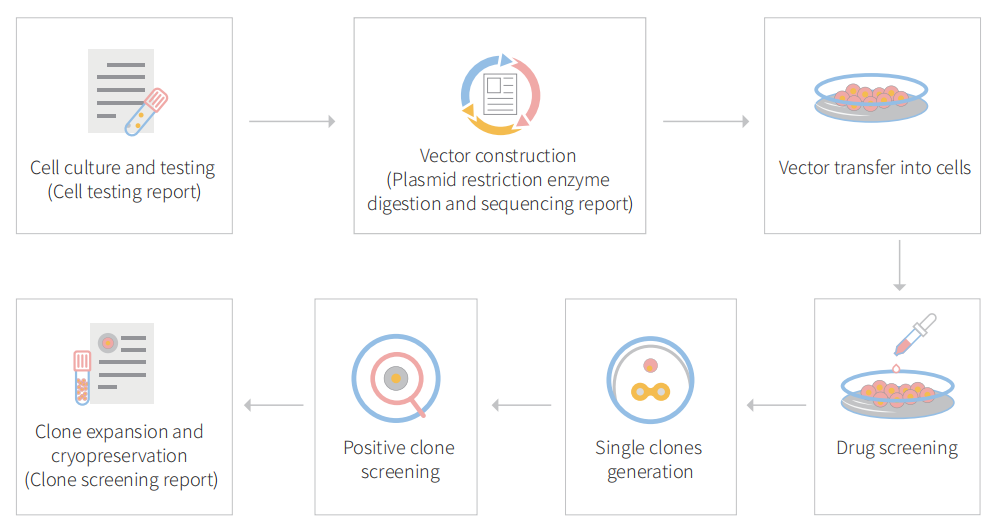
To accelerate ESCs and iPSCs research, Ubigene developed CRISPR-U™ for gene manipulation of ESCs and iPSCs. Thereby, possible to achieve genome editing in ESCs and iPSCs with the utilization of the CRISPR/Cas9 system. The CRISPR-U™ system used for rapid and precise gene editing in ESCs and iPSCs. CRISPR-generated cell models have enabled mechanism and treatment explorations of various diseases like cardiovascular diseases, liver diseases etc. In addition, CRISPR-generated cell models could be coupled with drug discovery, safety pharmacology and other therapeutic studies. Ubigene can customize the gene-editing in ESCs and iPSCs as well as can generate various genes modification in animal models.
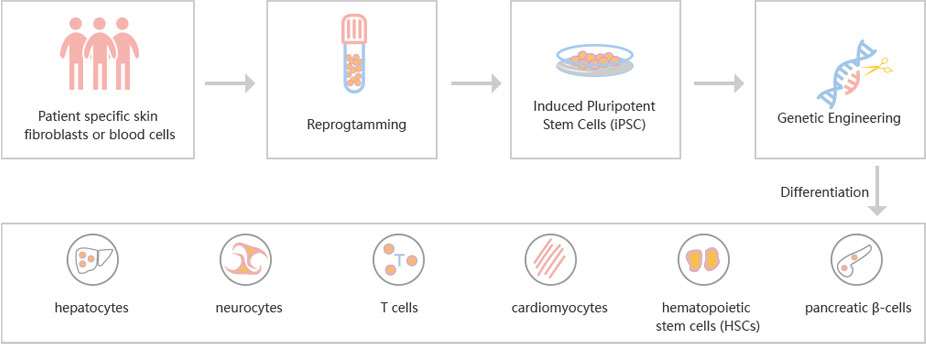
Figure: CRISPR-U™ customized workflow for engineered ESCs and iPSCs
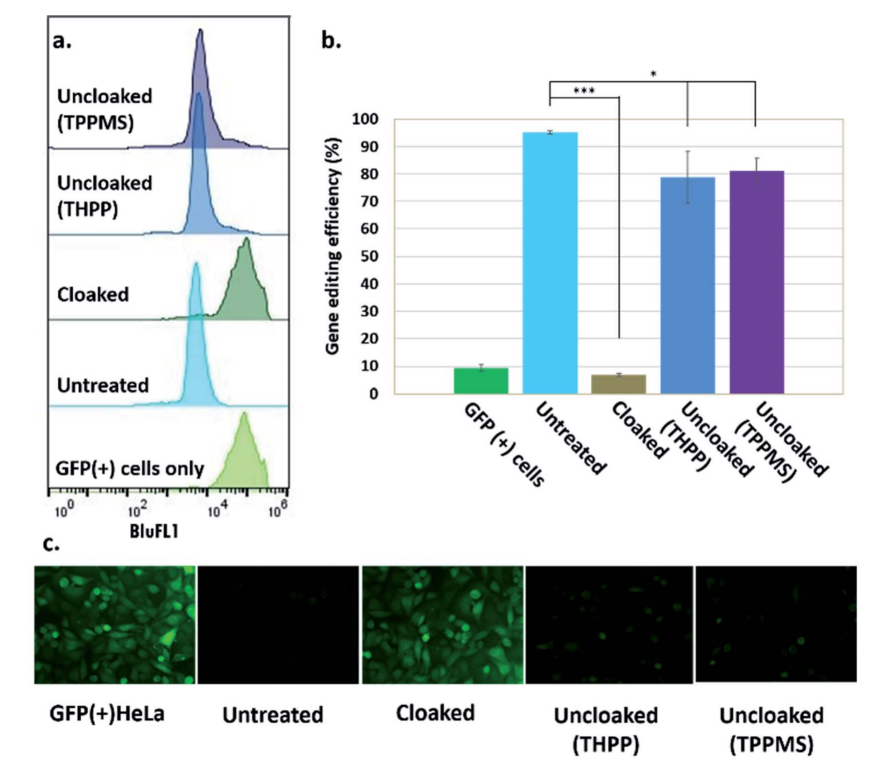
HLA Class I depleted platelet can evade natural killer cell immunity
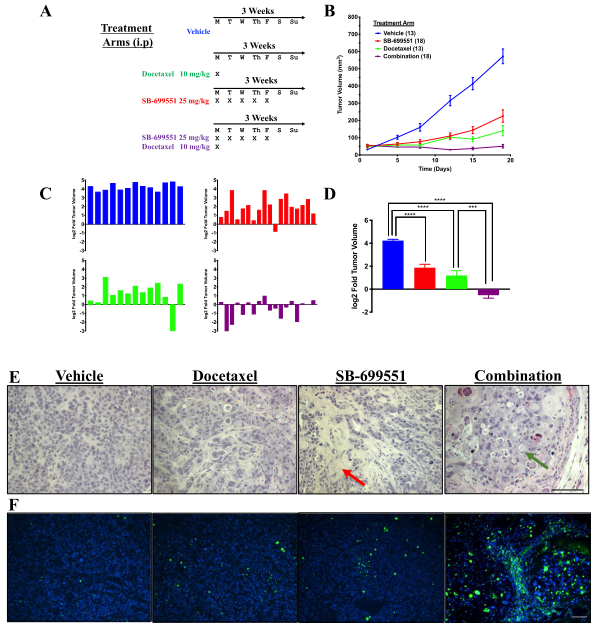
Platelet transfusion is an essential treatment for patients with thrombocytopenia. Alloimmune platelet transfusion refractoriness (allo-PTR) is observed in approximately 5%–15% of patients who receive platelet transfusion, with the most dominant cause being the production of alloantibodies against human leukocyte antigen class I (HLA-I). In such HLA-I-mediated allo-PTR, transfused platelets are immediately rejected, except for HLA-I-compatible platelets. However, the need to select compatible donors limits the supply, with the most difficult cases being rare HLA-I types. Human induced pluripotent stem cells (iPSCs) have been extensively studied as an ex vivo source for producing human cells and tissues and iPSC-derived platelets (iPLATs) have the potential to resolve the aforementioned issues in current transfusion systems. In the present study, CRISPR/Cas9 mediated HLA-KO iPLATs were generated by knocking out HLA-I complex molecule β2-microglobulin (B2M). Exon 1 of B2M was selected as the knockout target. For KO B2M, the researcher constructed sgRNA expression vector (pHL-H1-B2M-sgRNA-mEF1a-RiH), CRISPR/Cas9 expression vector (pHL-UbicP-SphcCas9-iP-A) and transfected MK-iPSCs to generate HLAKO iPLAT. HLA-KO iPLATs were deficient for all HLA-I but did not elicit a cytotoxic response by NK cells in vitro and showed circulation equal to wild-type iPLATs upon transfusion in humanized mice model (Hu-NK-MSTRG) reconstituted with human NK cells. This study revealed the unique non-immunogenic property of platelets and provides a proof of concept for the clinical application of HLA-KO iPLATs.
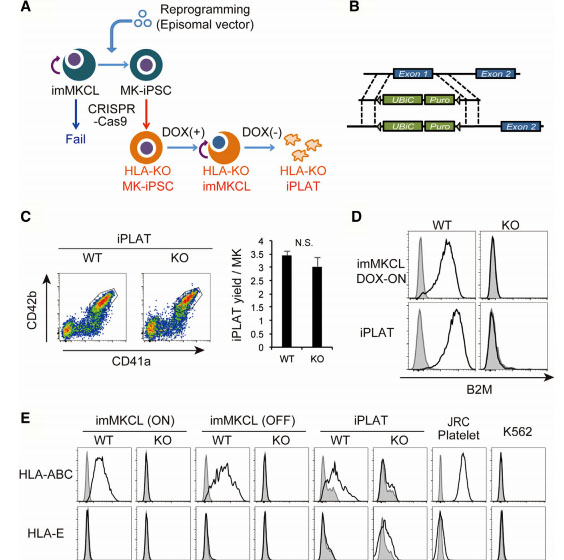
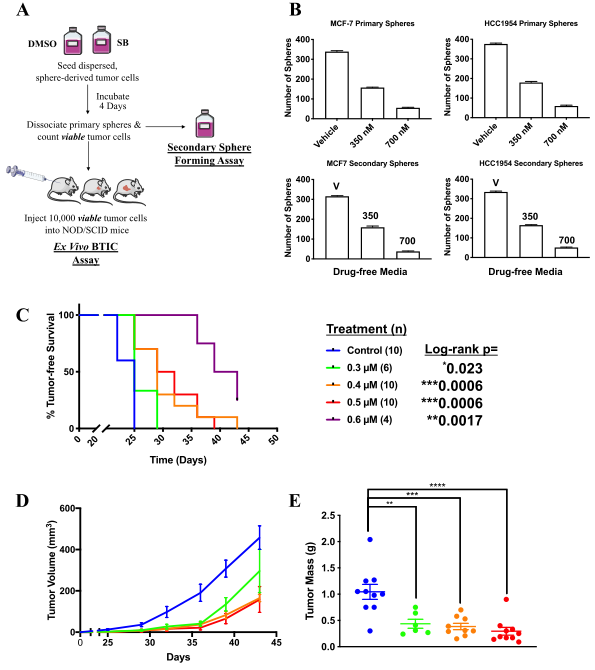
Fig1: Production of HLA-KO iPLATs by Knocking Out β2-Microglobulin in imMKCL
imMKCL was first re-reprogrammed to secondary iPSCs (MK-iPSC), in which B2M was knocking out. After expansion and matured MK-iPSCs release iPLATs contains Kocking out B2M (A, B). Flow-cytometry analysis of the generated CD41a+ CD42b+ iPLATs and the cell-surface expression of B2M and of HLA-ABC and HLA-E on imMKCLs, iPLATs, JRC platelets, and K562 cells (C, D and E).
Correction of recessive dystrophic epidermolysis bullosa using iPS cells
Dystrophic epidermolysis bullosa (DEB) is a rare genetic skin fragility disorder characterized by blistered skin and mucosa that can be inherited in either a dominant (DDEB) or recessive (RDEB) manner. DEB is caused by mutations in the COL7A1 gene that encodes type VII collagen (C7), a crucial protein that forms anchoring fibrils (AFs) that stabilize dermal-epidermal adhesion at the basement membrane zone (BMZ). Patients with RDEB lack functional C7 and have severely impaired dermal-epidermal stability, resulting in extensive blistering and open wounds on the skin that greatly affect the patient’s quality of life. There are currently no therapies approved for the treatment of RDEB. The researcher, used CRISPR system to modify induced pluripotent stem cells (iPSCs) derived from patients with RDEB in both the heterozygous and homozygous states and corrected mutations in exon 19 (c.2470insG) and exon 32 (c.3948insT) in the COL7A1 gene through homology-directed repair (HDR). They found that three-dimensional human skin equivalents (HSEs) were generated from gene-corrected iPSCs, differentiated into keratinocytes (KCs) and fibroblasts (FBs), and grafted onto immunodeficient mice, which showed normal expression of C7 at the BMZ as well as restored AFs 2-month post grafting. There findings represent a crucial advance for clinical applications of innovative autologous stem cell-based therapies for RDEB. This finding could serve as a foundation to translate this treatment into the clinic.
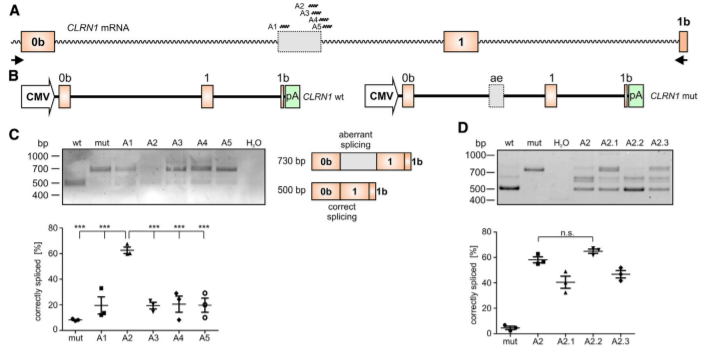
Biallelic Gene Correction of the Homozygous and Heterozygous Mutations in the COL7A1 Gene by CRISPR/Cas9 Plasmid approach and RNP approach:
The researchers employed two CRISPR-mediated genome editing approach for correction of COL7A1 gene mutation in homozygous and heterozygous state of iPSCs. They applied biallelic gene correction in homozygous iPSCs with CRISPR/Cas9 plasmid approach. They evaluate the efficacy of gene correction in exon 19 of COL7A1 gene in iPSC-derived cells. They found that 10% of the clones had undergone biallelic correction and 40% of clones had undergone monoallelic correction of the COL7A1 mutation in exon 19. The sequence analysis directed that the target sequence areas were free of off-target mutations. To improve correction efficiency, the researcher used the Cas9 protein and a chemically modified synthetic gRNA as a RNP complex. By using RNP approach, they achieved 58% biallelic and 42% monoallelic correction for the homozygous mutation (c.2470insG) in exon 19 of COL7A1, and 19% biallelic and 48% monoallelic correction for the heterozygous mutation (c.2470insG/ c.3948insT) in exons 19 and 32 of COL7A1, respectively. This strategy creates a “scarless” phenotype without leaving a residual footprint and generate a very high efficiency in single iPSCs without evidence of off-target activity.
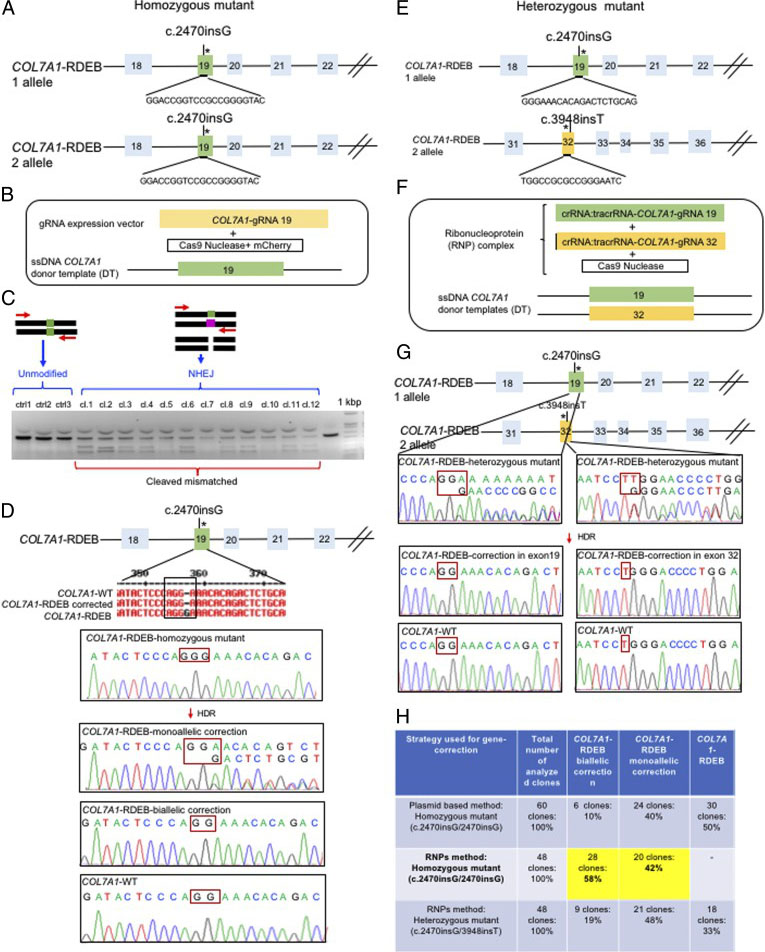
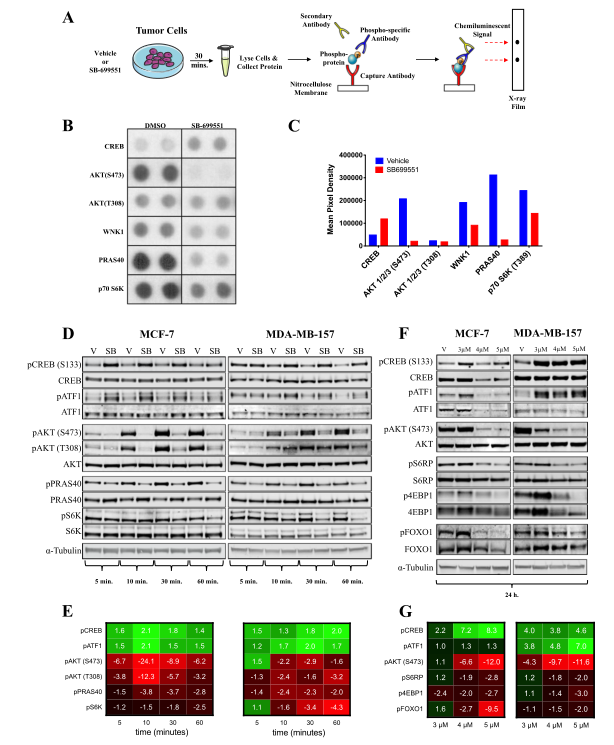
Fig. 2. Evaluation of CRISPR/Cas9 gene-correction efficiency using plasmid- and protein-based methods in iPS cells. (A) Schematic representation of the CRISPR target site for the homozygous (c.2470insG) mutation in exon 19 of COL7A. (B) Components used for plasmid-based gene-correction strategy. (C) T7E1 assay after gene editing targeting exon 19 of the COL7A1 gene. (D) Sanger sequencing confirmed various genotypes. (E) Schematic representation of CRISPR target site for heterozygous (c.2470insG/c.3948insT) mutations in exon 19 of COL7A. (F) Components used for protein-based gene-correction strategy. (G) Sanger sequencing confirmed various genotypes. (H) Summary of the detected genotypes and efficacy after CRISPR/Cas9 gene editing.
Skin integrity and type VII collagen restoration after grafting onto nude mice
The researcher established protocol for generating keratinocytes (iKCs) and fibroblasts (iFBs) from RDEB patient-derived iPSCs. They observed that functional iKCs need 60 days for maturation invitro and 96% of iPSC-derived KCs (iKCs) expressed keratin 14, and 50% of the 96% had high levels of p63 expression. Besides that, they obtained iFBs from iPSCs after 31 days differentiation. They observed that more than 90% of iPSC-derived iFBs with expression patterns consistent with those of normal human FBs. The 3D skin, derived from gene-corrected iKCs and iFBs, was then grafted onto immunodeficient mice and analyzed 2-month postgrafting. They analyze the expression of C7, epidermal differentiation markers such as keratin 14, keratin 10, loricrin, filaggrin, and vimentin. The iPSC-derived corrected xenografts expressed C7 and fully resemble the WT skin, demonstrating that the gene correction restored the protein function in iPSC-derived iKCs and iFBs.
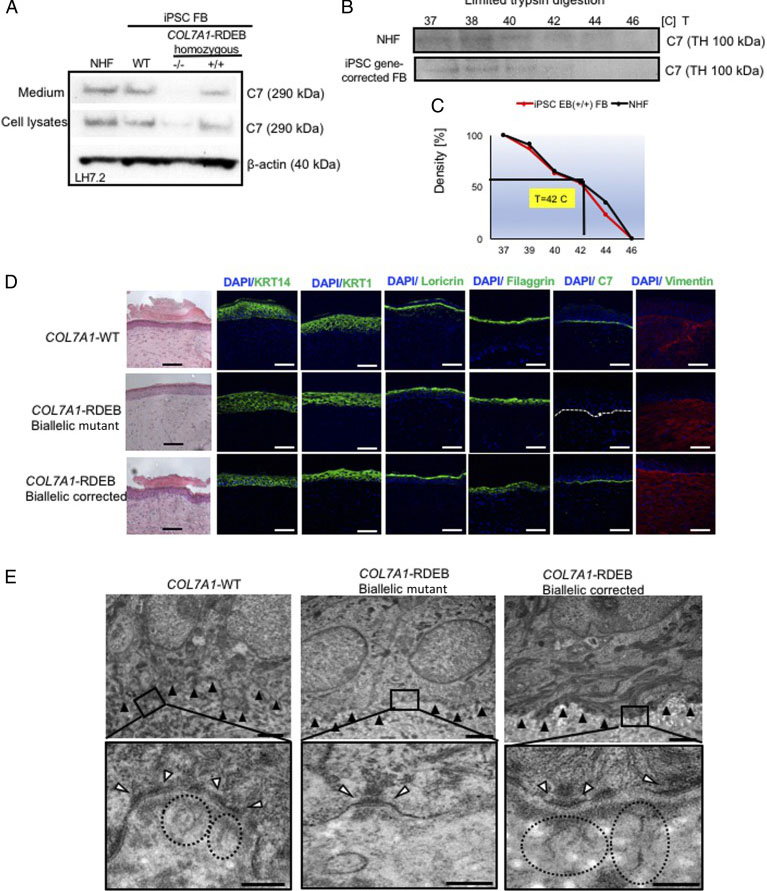
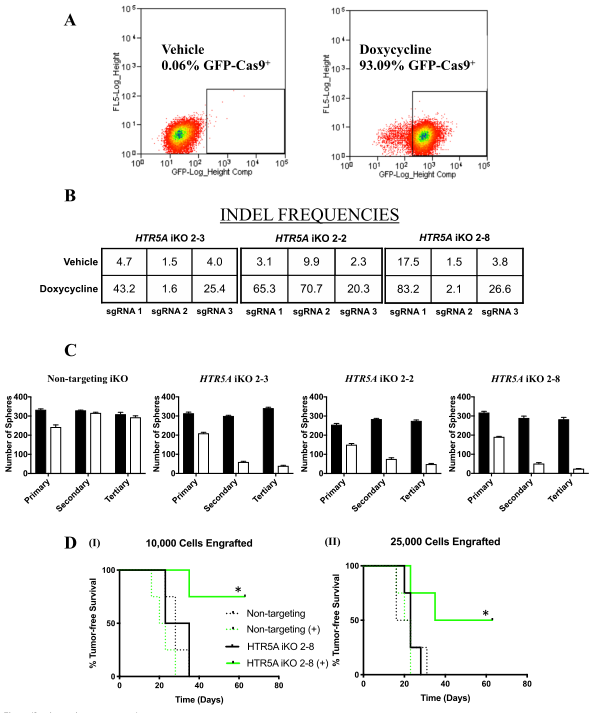
Fig. 3. Functional verification of gene-corrected RDEB patient iPSC-derived FBs and HSEs. (A) Western blot (WB) analysis assessing type VII collagen (C7) protein expression and secretion in normal human FBs, wild type iPSC-derived FBs (iPSC WT FB), COL7A1-RDEB homozygous mutant (−/−), and corrected (+/+) iPSCs differentiated to FBs. (B) Thermal stability of C7 was analyzed and quantified by (C) limited trypsin digestion of medium from normal human FBs and iPSC gene-corrected RDEB-derived FBs at increasing temperatures. (D) Generation of 3D HSEs using gene-corrected RDEB patient iPSC-derived KCs and FBs, which were then grafted onto nude mice are histologically comparable to those generated using iPSC WT KCs/FBs, 2-month post grafting. H&E staining revealed normal epidermal and dermal morphology. C7 deposition is demonstrated by immunofluorescence (IF) staining (green signal) 2 month after grafting, using LH7.2 antibody. Additional IF staining was performed for keratin 14, keratin 10, loricrin, filaggrin, and vimentin on corrected, mutant, and WT xenografts. (E) Transmission electron microscopy was performed on positive iPSC WT KCs/FBs skin grafts, negative COL7A1-RDEB homozygous mutant and COL7A1-RDEB gene-corrected KCs/FBs skin grafts, and gene-corrected RDEB skin grafts, 2-month post grafting. The BMZ is indicated by black arrowheads.
A novel autologous cell therapy for LGMD2A
Limb girdle muscular dystrophy type 2A (LGMD2A) is an autosomal recessive inherited disorder and the most common form of LGMD. LGMD2A occurs due to loss of functional Calpain 3 (CAPN3), a skeletal muscle-specific isoform of the calcium-sensitive Calpain cysteine protease family. The researcher used CRISPR-Cas9 mediated genome editing to iPSCs from three LGMD2A patients to enable correction of mutations in the CAPN3 gene. A CRISPR mediated gene knockin approach were used to edit iPSCs genome carrying three different CAPN3 mutations, and rescue CAPN3 protein in myotube derivatives in vitro. To corrected the mutation, the researcher used guide RNA sequence that targeting CAPN3-exon 14 (50 -CATCTCCGTGGATCGGCCAG-30) cloned in pX458 vector. HDR donor vector was constructed in the pBluescript plasmid backbone with selection cassette of loxP-flanked GFP-2A-neoR driven by HEF1-eIF4g for positive selection and HSV-tk driven by MC1 promoter for negative selection. The 5 -prime homology arm consisting of intron 13 (950 bp), the knockin insert consisting of exons 15–24 cDNA, and sv40 poly(A) signal sequence were in upstream of GFP-2A-neoR cassette. The 3-prime homology arm consisting of the In14 (950 bp) were downstream of the GFP-2A-neoR cassette. To test the rescue of CAPN3 expression in vivo, transplanted gene-corrected iPSC-derived myogenic progenitors into cardiotoxin-pre-injured tibialis anterior (TA) muscles of C3KO-NSG mice and rescue of the CAPN3 mRNA.
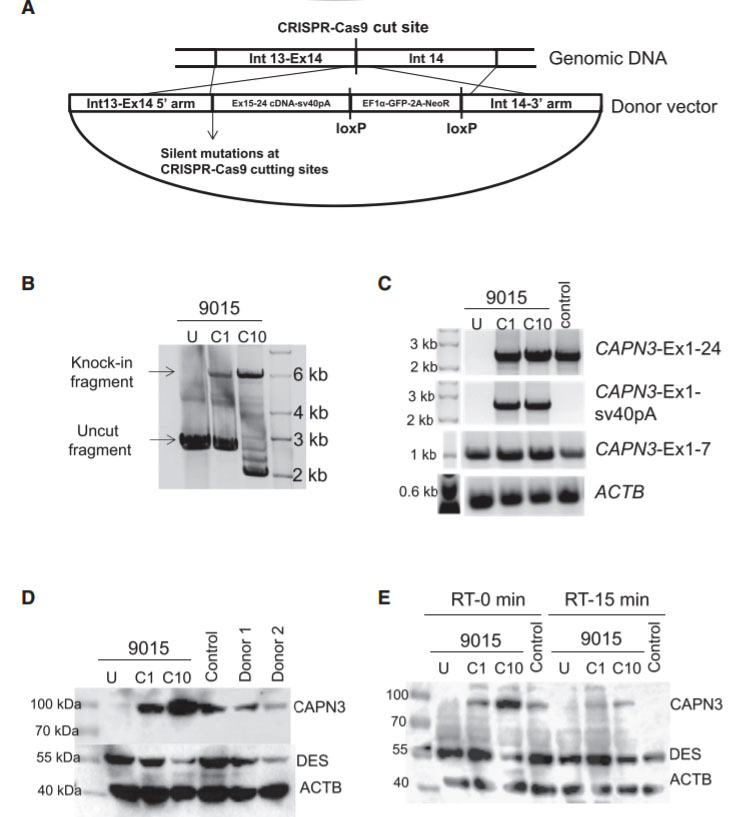
Fig 4: Gene Correction of CAPN3 Mutation in 9015 LGMD2A iPSCs
In this study, the researcher employed a homology-directed repair for gene knockin-based correction of CAPN3 mutations (A). LGMD2A iPSCs gene corrected (C1 and C10) spanning region amplified by genomic PCR. RT-PCR analysis were performed of gene-corrected and uncorrected 9015 iPSC-derived myotubes as well as of unaffected myotubes (control). Rescue of CAPN3 protein expression in gene-corrected (C1 and C10) LGMD2A iPSC-derived myotubes were performed with western blot. In the validation study, the patient’s parents and unrelated control iPSC-derived myotubes were used as reference (D). In addition, autocatalytic activity of CAPN3 were also analyses by western blot, the lysates obtained from myotubes incubated for 0 or 15 min at room temperature (E).