Salmonella is a facultative anaerobic gram-negative rod-shaped bacteria. Salmonella belongs to Enterobacteriaceae and is an important medical pathogen of humans and animals. Salmonella forms a complex bacterial community consisting of two species and six subspecies, including more than 2579 serotypes. At present, there are two species of Salmonella, S. entreica and S. bongori. The selective targeting of tumor tissue by Salmonella also makes it an ideal vector for tumorAs an intracellular parasite, Salmonella can effectively replicate and inhibit tumor growth in tumor tissue. After genetic engineering, it can be used as a carrier of tumor gene therapy in vitro and in vivo for liver cancer, gastric cancer and colorectal cancer.

Salmonella and Tumor Therapy
1. Direct antitumor therapy with attenuated Salmonella
In view of the characteristics of Salmonella that can effectively inhibit tumor growth, scientists use a variety of genetic engineering techniques to modify the chromosome genome of Salmonella, which can reduce the virulence of Salmonella, so as to obtain the attenuated strain. While reducing the pathogenicity to the host, it still keeps high immunogenicity, thus ensuring the safety of clinical application.
2.Tumor targeting gene therapy
Attenuated Salmonella can carry exogenous genes, cytokines and exogenous effector proteins to treat tumors. Cytokines play an anti-tumor role by killing tumor cells directly. Exogenous effector proteins can be effectively transferred by attenuated Salmonella and expressed therapeutic proteins
Knockout PhoP to construct attenuated Salmonella, improves the safety of Salmonella in tumor treatment
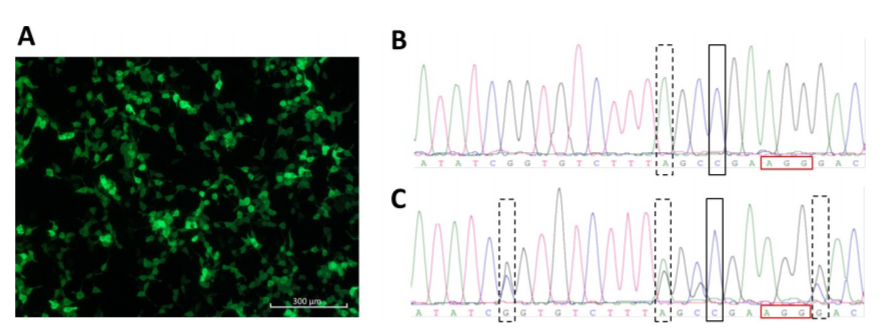
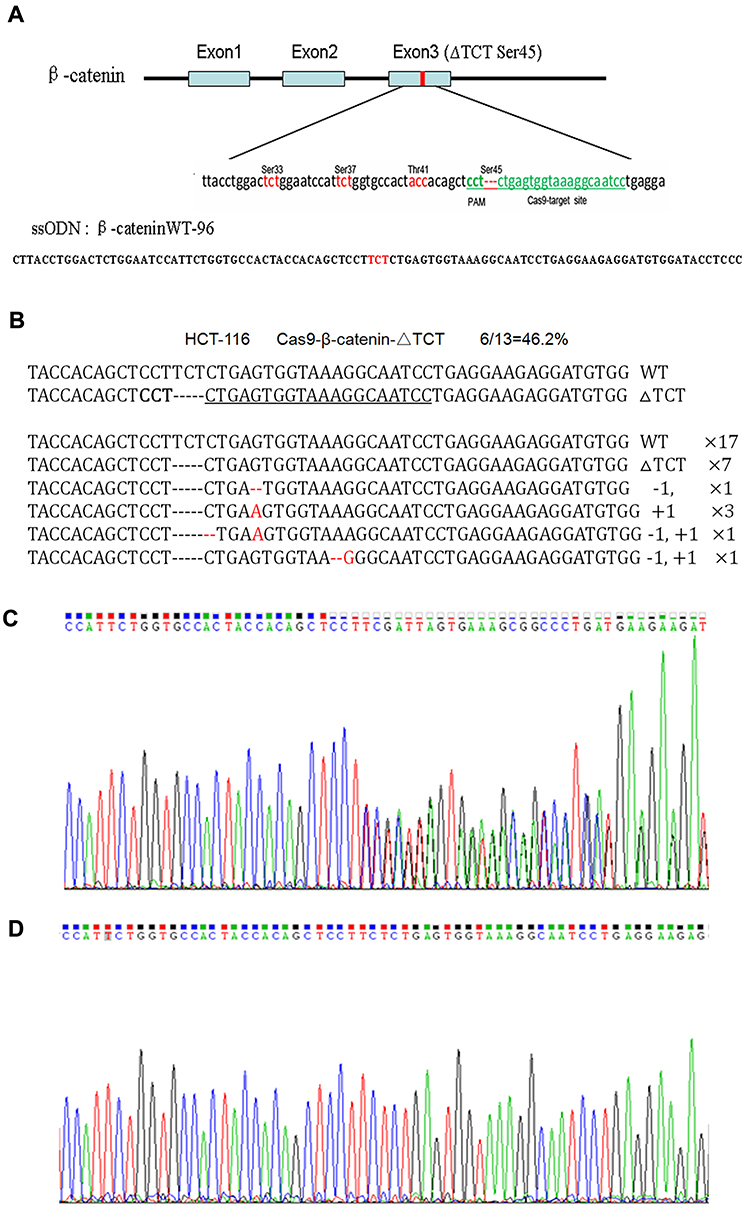
The scientists used gene-splicing PCR combined with λ-Red system to delete PhoP from wild-type Salmonella typhimurium. phoP is a transcription regulator and a component of two regulatory systems that play key roles in adaptation to the homeostelium and in macrophage survival. It also controls the expression of more than 40 genes required by Salmonella typhimurium and its resistance to adverse environments in the host, such as low pH in the stomach, bile salt, hypoxia in the small intestine, and cationic antimicrobial peptides on epithelial cells. The disruption of phoP of Salmonella typhimurium leads to its inability to survive in phagocytes, and increases its sensitivity to stress, so as to achieve the function of reducing toxicity, and develop a safer bacterial therapy.

The phoP of wild type and standard strain of Salmonella typhimurium was disrupted by SOEing PCR. Three standard PCR and one fusion PCR were used to construct the linear DNA containing the upstream and downstream of phoP sequence and kanamycin cassette. pKD4 carrying kanamycin flanked by FRT (FLP recognition target) was used as a template plasmid. PCR confirmed that kanamycin had replaced phoP gene. Therefore, attenuated Salmonella was successfully constructed by removing phoP gene.
Gene-editing bacteria bring infinite possibilities to research, clinical medicine agriculture and other industrial productions!
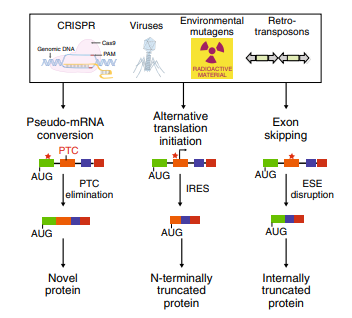
Gene knockout has been used for many purposes, such as studying gene function, vaccine production and improving protein structure or expression. At present, the widely used traditional gene-editing methods include R6K suicide plasmid and λ-Red system.
Although the λ-Red system seems simple and has been successfully applied to E.coli and other gram-negative bacteria, the performance of this system in different bacteria is unstable due to the inherent differences of bacteria. In addition, suicide plasmid has some disadvantages such as host limitation and resistance residue. Other shortcomings of traditional KO include low recombination efficiency, heavy workload on validation, and residual loxP or FRT sites.
CRISPR/Cas9 technology is the most rapidly developed gene-editing technology in recent years. However, due to the lack of a repair system in bacteria, only a few microbial species were published to modify genes with this technology.Ubigene developed CRISPR-B™ technology, which is highly efficient in genome editing. CRISPR-B™ has features of easy-to-handle, accurate targeting, low off-target effect, scarless. It is efficient in various bacteria. The efficiency is more than 20x higher than that of traditional methods, easily achieve gene knockout, point mutation and knockin.
A new treatment for tumor--CTNNB1-shRNA expression in Salmonella
Bacterial therapy has been used clinically for decades with a proven track record of safety in the treatment of gastroenteric diseases (for example, diarrhea, irritable bowel syndrome and inflammatory bowel disease). Moreover, recently there has been renewed interest in the clinical applications of live bacteria which is various non-pathogenic anaerobic bacteria are given intravenously and can infiltrate and replicate within solid tumors, particularly to treat human solid tumors. RNA interference (RNAi) has been established as an important research tool with great potential for gene therapy. Using a combination of bacterial therapy and RNAi therapy, scientists developed bacteria-mediated RNAi that delivers shRNA-expressing vectors to target cells to silence disease-causing genes.

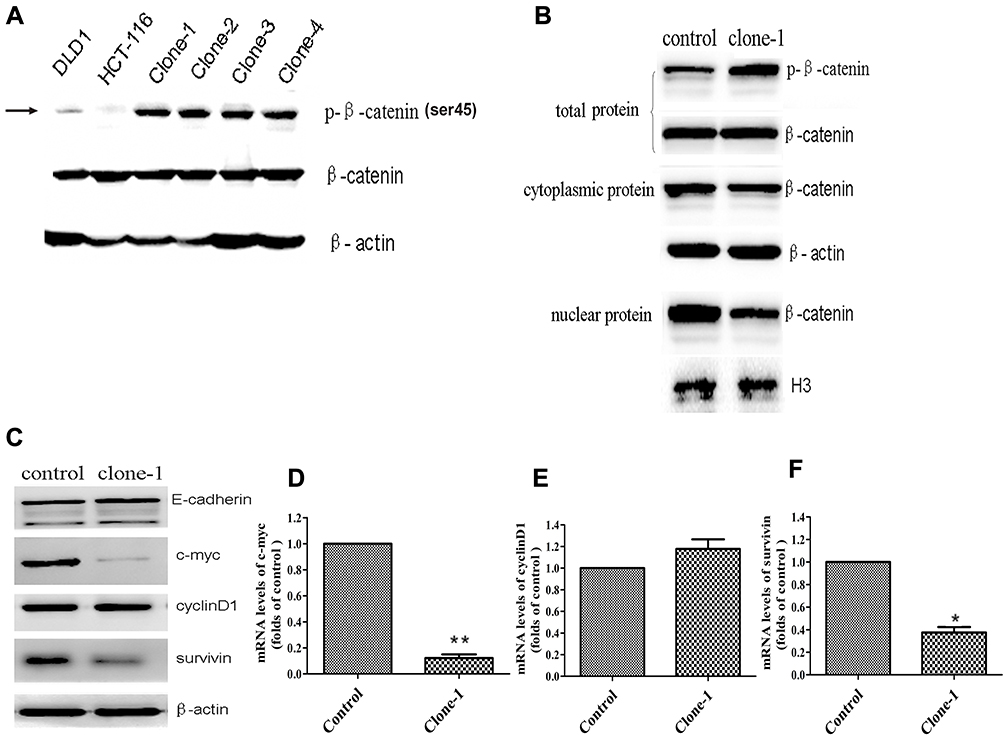
Scientists have established a pSLS plasmid system that can express shRNAs in attenuated Salmonella. shRNA against HIV tat gene, human and mouse CTNNB1 genes were inserted into the multiple cloning site of pSLS. Then these plasmids were transformed into attenuated Salmonella, resulting in corresponding SL-pSLS-TAT, SL-pSLS-huCAT and SL-pSLS-mCAT strains. To determine whether SL-PSLS-HucAT can knockdown CTNNB1 expression of SW480 and inhibit the growth of SW480, SW480 cells were treated at different MOI.
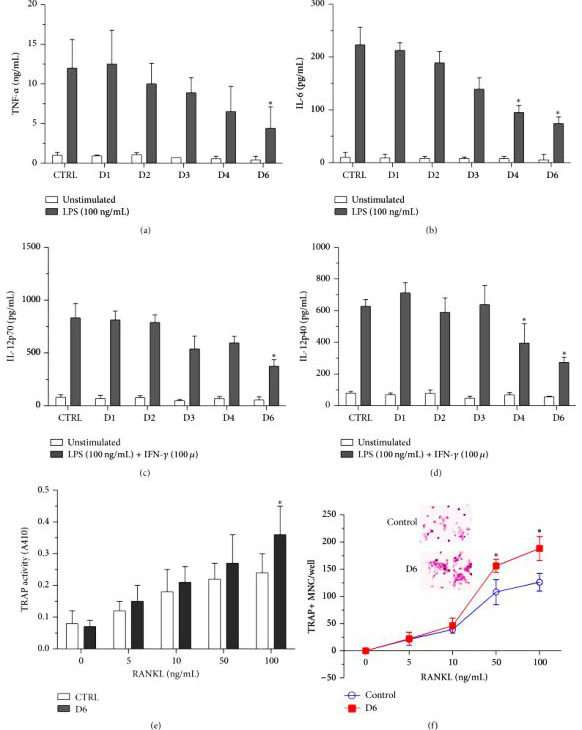
CTNNB1 gene silencing significantly reduced cell proliferation and death in SW480 cells compared with control cells.To determine whether shRNA-expressing Salmonella could mediate anticancer effects in vivo, researchers performed studies using the SW480 xenograft tumor model. For these experiments, BALB/c female nude mice with established SW480 xenograft tumors were randomized into three groups to receive phosphate-buffered saline (PBS), SL-pSLS-TAT or SL-pSLS-huCAT. Tumor growth, as well as the expression levels of CTNNB1 and its downstream target genes c-Myc and cyclin D1, were recorded over a period of 2 weeks. Significantly, the growth of xenograft tumors treated with SL-pSLS-huCAT was reduced by 65% compared with tumors from the PBS control group, and by 45% compared with tumors treated with SL-pSLS-TAT . Reduced c-Myc and cyclin D1 protein levels were also observed in tumors treated with SL-pSLShuCAT compared with those treated with PBS. Thus,a significant reduction in tumor CTNNB1 protein level was observed when the mice were treated with SL-pSLS-huCAT.These results revealed that SL-pSLS-mCAT reduced the mRNA level of CTNNB1 in small intestines (by 34%), in polyps (by 73%) and in mucosal tissues (by 83%,) when compared with SL-pSLS-TAT-treated mice. The greater reductions observed in polyps and mucosal tissues over those found in the small intestines suggest a more selective invasion of these former tissues by Salmonella.
Taken together, these data suggest that attenuated shRNA-expressing Salmonella may be a powerful new tool for in vitro gene silencing, functional genomics, and the development of RNAi-based anticancer or human immunodeficiency virus therapeutics.
Ubigene developed CRISPR-B™ which optimizes the microbial gene-editing vectors and process. The efficiency and accuracy are 20x higher than traditional methods. CRISPR-B™ can be used in gene editing of bacteria and fungi. Contact us immediately to know about your research related services!
Reference:
1. A.Andino and I. Hanning. Review Article Salmonella enterica: Survival, Colonization, and Virulence Differences among Serovars. 2015. e Scientifific World Journal.
2. Zhu Xiaozhou, Kong Guimei, Wan Dan, sun Guozhuang, Jiao Hongmei, Yin Yinyan, Li Guocai. Research progress of attenuated Salmonella in digestive system tumors. 2017. World Chinese Journal of digestion. 1480-1485
3. H Guo, J Zhang and C Ina. Targeting tumor gene by shRNA-expressing Salmonella-mediated RNAi. Gene Therapy. 2011. 18:95-105
4. Ahani Azari, A. Zahraei Salehi, T. Nayeri FasaeiB.and Alebouyeh, M..Gene disruption in Salmonella typhimurim by modified λ Red disruption system. Iranian Journal of Veterinary Research ,Shiraz University. 2015. 52:301-305