Human Genome Project and DNA sequence data obtained from diseased individuals have provided an unprecedented opportunity for understanding genetic components allied with human diseases. Mutations in cancers likely alter a great number of molecular events. One of these events is protein phosphorylation, which when altered may result in system-wide disruption and deregulation of signal transduction.
To date, more than 3000 altered human genes are known to be associated with diseases. Monogenic disorders, such as Huntington's disease, cystic fibrosis, thalassemia, and sickle cell anemia, are caused by single-gene mutations while multifactorial diseases such as cancer and diabetes resulted from an interplay between numerous genetic mutations and environmental conditions. There are many known spontaneous or somatic amino acid substitutions and some of these will likely have profound effects on protein function. Within these sets of mutations, we expect to observe loss-of-function mutations that turn-off normal molecular function. We also expect to observe mutations that cause a gain of function; that is, mutations that cause a molecular function to have deregulated activation when compared to normal function. Finally, we expect to observe many mutations that do not participate in neoplastic development and progression.
Phosphorylation of amino acid residues serine (S), threonine (T), and tyrosine (Y) are common in cancer-associated proteins and known to be deregulated in cancer. It is well understood that changes in phosphorylation signaling can be due to deregulation of kinase and phosphatase function, usually detected through altered gene expression. Amino acid substitutions on kinases or phosphatases directly interrupt the stability and/or the function of the kinase or phosphatase, resulting in changes in target phosphorylation. Effects of kinase or phosphatase regulators can also lead to altered phosphorylation. Disruptions of phosphorylation sites are associated with cancer, for instance, mutations of T286 in cyclin D1 (CCND1). Phosphorylation of T286 by GSK3B in the wild type form of cyclin D1 initiates its nuclear export and subsequent degradation in the cytoplasm, while the loss of phosphorylation is causatively implicated in nuclear accumulation of cyclin D1 in esophageal cancer and generally increased oncogenic potential. In another study in catenin β-1, which is involved in hepatocarcinoma, phosphorylation is known to occur on amino acids T41 and S45 and result in significant scores for loss-of-phosphorylation. Thus, phosphorylation target site mutation is a novel route for cancer development. A mechanism of signal transduction deregulation in cancer is mediated by either removal or creation of phosphorylation sites thereby causing either a loss or a gain of phosphorylation function, depending on the role of the phosphorylated residue. Phosphorylation disrupting mutations, we can find several pathways are enriched in mutations. Notably, the Wnt/β-Catenin pathway is enriched in both the gain and loss of phosphorylation sites.
Phosphorylation can affect protein functions in various ways, like increase or decrease a protein's activity, stabilize it or mark it for destruction, localize it within a specific cellular compartment, and it can initiate or disrupt its interaction with other proteins. More than two-thirds of the 21,000 proteins encoded by the human genome is phosphorylated, and, likely, more than 90% are subjected to PTMs. Phosphorylation events, therefore, play an important role in the control of biological processes such as proliferation, differentiation, and apoptosis. The phosphorylation sites of a protein often have key regulatory functions and are central to many cellular signaling pathways, so mutations that modify them have the potential to contribute to pathological states such as cancer, cardiovascular disease, renal disease, etc. However, the regulatory importance of phosphorylation sites can vary greatly. Mutations in the phosphorylation sites have the potential to greatly influence protein structure and function by up- or downregulating the stoichiometry of phosphorylation at that site. Furthermore, mutated phosphorylation frequently modifies protein-protein interaction (PPI) partners and this modification is central to many cellular signaling pathways, another important consideration is the impact of disrupting or altering phosphorylation sites on its PPI, and indeed on the whole PPI network. The study of molecular interaction networks has become increasingly applied to human diseases such as cancer, with the effect of many known cancer mutations being understood through their effect on PPI networks.
The potential area of protein phosphorylation study on a system level.
1. Basic Research: Study on phosphorylation regulation profile (molecular fingerprint to identify related and opposing signaling states).
2. Disease Modeling: Use of gene manipulation tools to construct disease model for cells and animals (Study of the functional analysis of targeted mutations in disease-related genes in the isogenic background, Study the pathology of diseases and their development)
3. Cell/gene-Based Therapies: Genomic medicine as diseases associated with defective genes can be rectified at the level of the genome itself (Gene editing for human therapy & cell therapy)
4. Drug Discovery: Phosphorylation sites within cellular proteins and their regulatory role allow to use of phosphorylation as an assay endpoint and obtained information can be used at various stages of the drug discovery and development processes.
As techniques providing precise, targeted modifications and correction to genome sequences, genetic medicine proves to have extensive promise as a therapeutic intervention against human diseases. Furthermore, using CRISPR/Cas9 technologies, editing of the genome is becoming straightforward so a 'knock-in' point mutation of a phosphorylation site can be more efficiently prepared and analyzed in vivo as well as in-vitro.
Study of monogenetic liver disease by the CRISPR system
Monogenetic liver disease, Wilson's disease (WD) is arises based on a mutation of the ATP7B gene and leads to a functional deterioration in copper (Cu) excretion in the liver. The excess Cu accumulates in various organs such as the liver and brain. Patients with WD show clinical heterogeneity, which can range from acute or chronic liver failure to neurological symptoms. The course of the disease can be improved by a life-long treatment with zinc or chelators, but serious side effects have been observed in a significant portion of patients, e.g. neurological deterioration and nephrotoxicity, so that a liver transplant would be inevitable. An alternative therapy option would be the genetic correction of the ATP7B gene. In this study the researcher, introduce artificial ATP7B point mutation in a human cell line using CRISPR/Cas9 gene editing, and corrected this mutation by the additional use of single-stranded oligo DNA nucleotides (ssODNs), simulating a gene correction of a WD point mutation in vitro.

Fig1: Generation of ATP7B knockout mutant
CRISPR/Cas9-mediated ATP7B KO cells were generated with reduced Cu resistance. HEK293T was used to generate the KO model and cells were lipofected with PX459. Cellular metabolic activities of cells were determined by MTT assay from both HEK293T & KO mutant (Fig1A). Sanger sequencing confirmed that HEK293T cell clone #1 carrying after deletion of one cytosine nucleotide at position 1184 (black rectangle), three nucleotides upstream the PAM region (red rectangle).
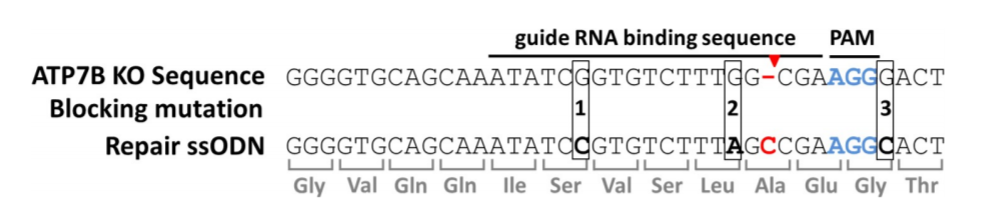
Fig 2. Xenotransplant of WT and TERT+/− Hela cells in nude mice
ATP7B KO (HEK293TΔC) cells exhibit a point mutation in form of a cytosine nucleotide deletion (red hyphen). Cas9 cuts three nucleotides (red arrow) upstream of the PAM region (blue). Repair ssODNs contain silent blocking mutations at positions 1, 2 and 3 (ssODN_3M) or at positions 2 and 3 (ssODN_2M).
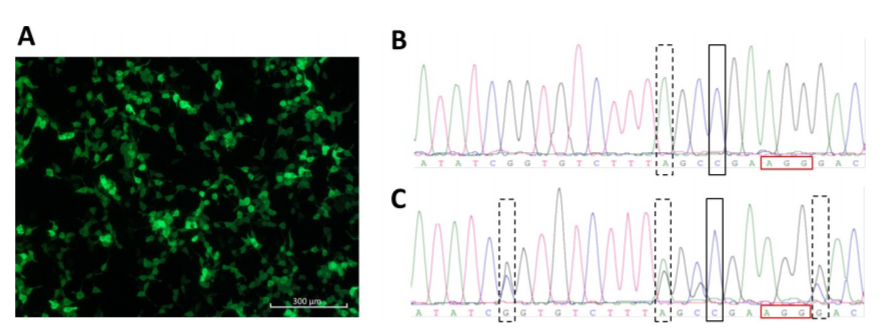
Fig 3. CRISPR/Cas9-mediated ATP7B repair.
HEK293TΔC cells were lipofected with pmaxGFP. GFP expression is shown in (Fig 3A). Sanger sequence analyzes of an ATP7B repaired homozygous HEK293TΔC cell clone (rectangle) carrying blocking mutation no. 2 (dashed rectangle) and of an ATP7B repaired heterozygous HEK293TΔC cell clone (rectangle) carrying all three blocking mutations (dashed rectangles) (Fig3 B, C).
These findings providing evidence that CRISPR/Cas9-mediated correction of ATP7B point mutations is feasible and may have the potential to be transferred to the clinic.
Correction of β-catenin ΔTCT Ser45 deletion mutation in HCT-116 Cells using CRISPR/Cas9
Colorectal cancer is the third most common cancer. CRC has a strong association with the deregulation of Wnt/β-catenin signaling pathway. β-catenin is phosphorylated at Ser45 by casein kinase 1 (CK1), and consecutively at Ser33, Ser37, and Thr41 by glycogen synthase kinase-3β (GSK-3β), which trigger the subsequent ubiquitination and proteasomal degradation. Mutations of these Ser/Thr residues alters functionally significant phosphorylation sites, inhibit the β-catenin phosphorylation-degradation cascade, and results in constitutive activation of Wnt/β-catenin signaling pathway. Then, correcting β-catenin genetic mutation by gene-editing technology may develop a next-generation therapeutic approach for colon cancer.
The human colorectal cell line HCT-116 was chosen for this study as it has a heterozygous deletion mutation (ΔTCT) of β-catenin genes, which is responsible for encoding the regulatory 45 serine (Ser45) at the N-terminal region of the protein. The researcher confirmed the location of ΔTCT Ser45 deletion mutation on the exon 3 of β-catenin gene and the CRISPR/Cas9 PAM site by gene sequencing. A 96-nt ssODN containing the wild-type β-catenin gene sequence as a template for HDR repair. After HCT-116 cells were co-transfected with the vector and ssODN, GFP-positive cells were sorted by FACS and expanded, and the mutation correction efficiency was calculated by TA cloning and sequencing.
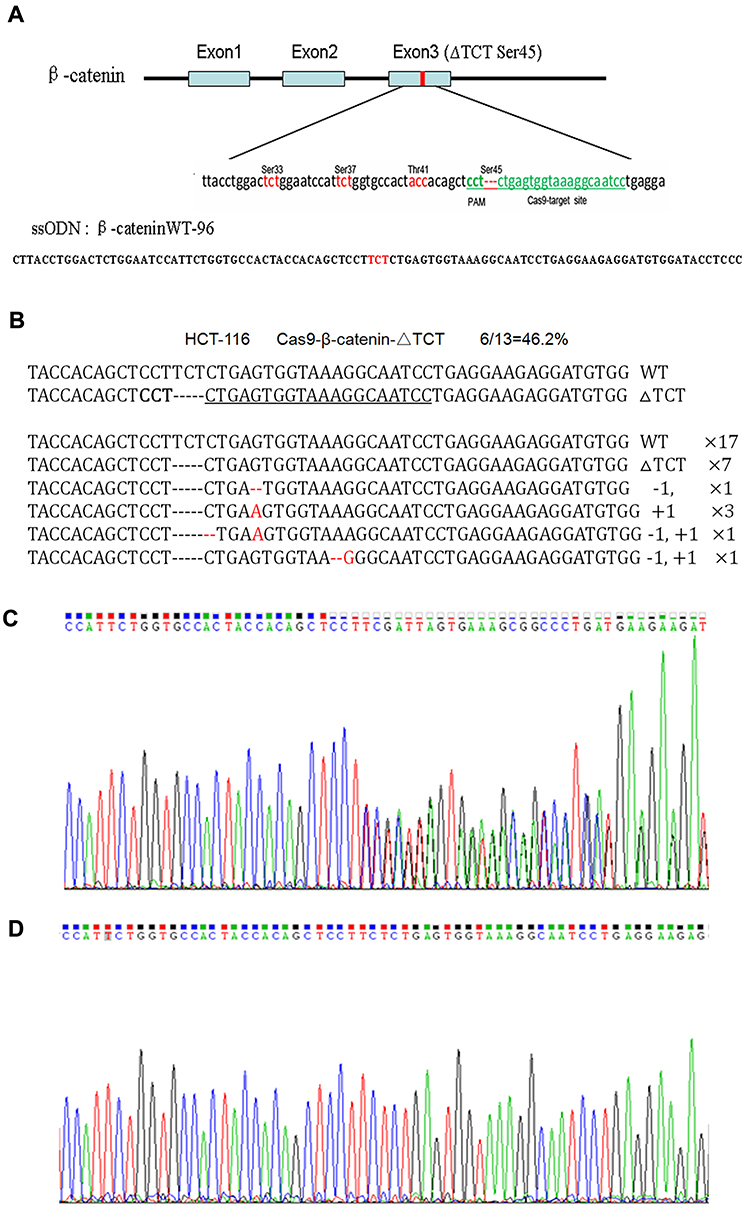
Fig 1: Mutation correction of β-catenin ΔTCT Ser45 in HCT-116 cells.
Due to a deletion mutation in one allele of the gene, the sequencing map of β-catenin in HCT-116 cells showed overlapped peaks starting from the mutation locus (Fig 1C). After mutation was corrected, we found the presence of a TCT sequence in the mutation locus, and the overlapped peaks starting from the mutation locus was disappeared accordingly (Fig 1D). These findings confirmed that co-transfected with the Cas9-GFP/sgRNA co-expression vector and ssODN could correct the β-catenin ΔTCT Ser45 deletion mutation in HCT-116 cells.
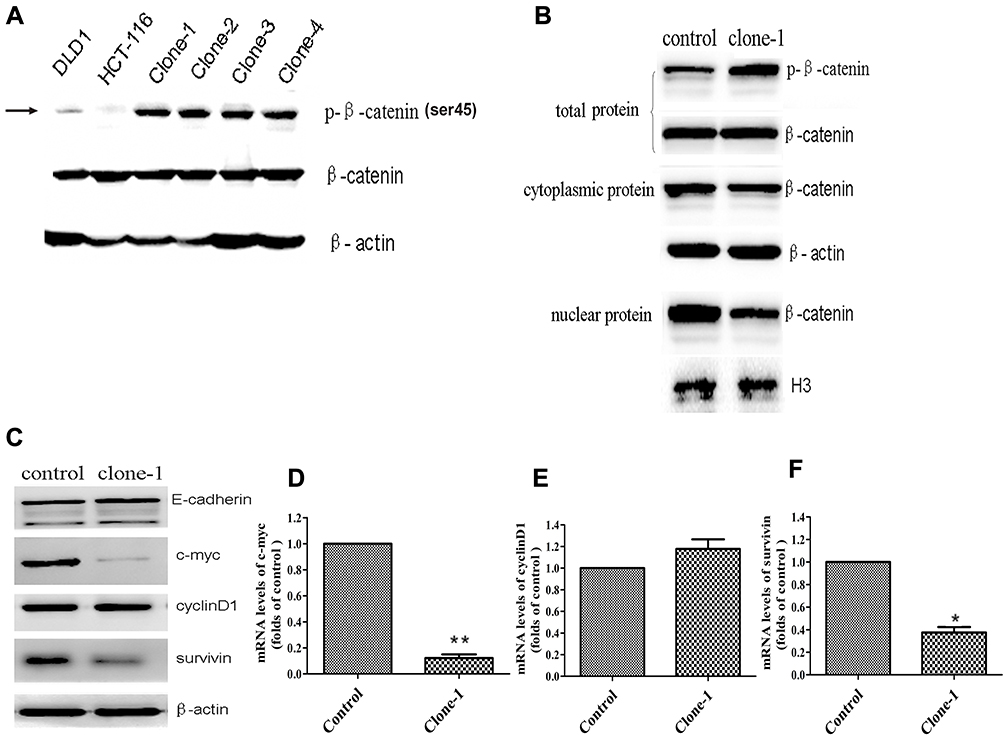
Fig 2: Functional effect of β-catenin ΔTCT Ser45 mutation correction
Ser45 of the β-catenin is an important phosphorylation site. Deletion mutation of the codon ΔTCT will result in a significant effect on β-catenin Ser45 phosphorylation in HCT-116 cells. Therefore, the researcher study on β-catenin expression levels in both deleted mutant and corrected mutant. As expected, uncorrected HCT-116 cells had very weak detectable Ser45 phosphorylated β-catenin expression, compared to the DLD1 colorectal cancer cells without β-catenin ΔTCT deletion mutations (Fig2A). They also studied the protein expression namely total, nuclear, and cytoplasmic β-catenin levels in the control and one mutation-corrected HCT-116 cell clone (Fig2B). Besides, they compare the expression of protein as well as genes of E-cadherin, c-myc, cyclinD1, and survivin gene (Fig 2C, D, E, and F).
In summary, correction of the driven mutation by the combination of CRISPR/Cas9 and ssODN could greatly remedy the biological behavior of the cancer cell line, suggesting a potential application of this strategy in gene therapy of cancer.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-
editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

No comments:
Post a Comment